


ANNA SOLINI (1), EDOARDO VITOLO (1), MARIO LUCA (2) MORIERI, GIOVANNI ZULIANI (2)
http://www.sisa.it/upload/GIA_2012_n2_9.pdf
(1) Dipartimento di Medicina Interna Università di Pisa; (2) Dipartimento di Medicina Clinica e Sperimentale Università di Ferrara; Sezione di Medicina Interna, Gerontologia e Nutrizione Clinica
Sommario
L’infiammazione è meccanismo fondamentale nel determinismo dell’aterosclerosi. Il cluster di fattori di rischio cardiovascolare che configura la sindrome metabolica è caratterizzato da insulino resistenza ed aumento della adiposità viscerale. Negli ultimi anni è stato dimostrato come il tessuto adiposo sia un vero e proprio organo endocrino in grado di produrre e rilasciare numerosi mediatori infiammatori, globalmente definiti come adipochine, che svolgono un ruolo rilevante nell’omeostasi energetica, nella modulazione della risposta infiammatoria tissutale e della risposta immune e nei processi angiogenetici.
Recenti evidenze suggeriscono anche un ruolo rilevante della microflora intestinale nel controllo del metabolismo energetico, dello stato infiammatorio generale e della plasticità del tessuto adiposo, con i recettori toll-like (TLRs) identificati come possibile anello di congiunzione tra sistema immune, risposte ormonali e alterazioni metaboliche.
In questo articolo vengono schematicamente descritti alcuni meccanismi attraverso cui l’infiammazione può indurre insulino resistenza, con particolare riguardo al ruolo del tessuto adiposo viscerale e del sistema immune innato, ripercorrendo le principali tappe del signaling intracellulare che mediano la complessa interazione tra reazioni infiammatorie e insulino resistenza sistemica.
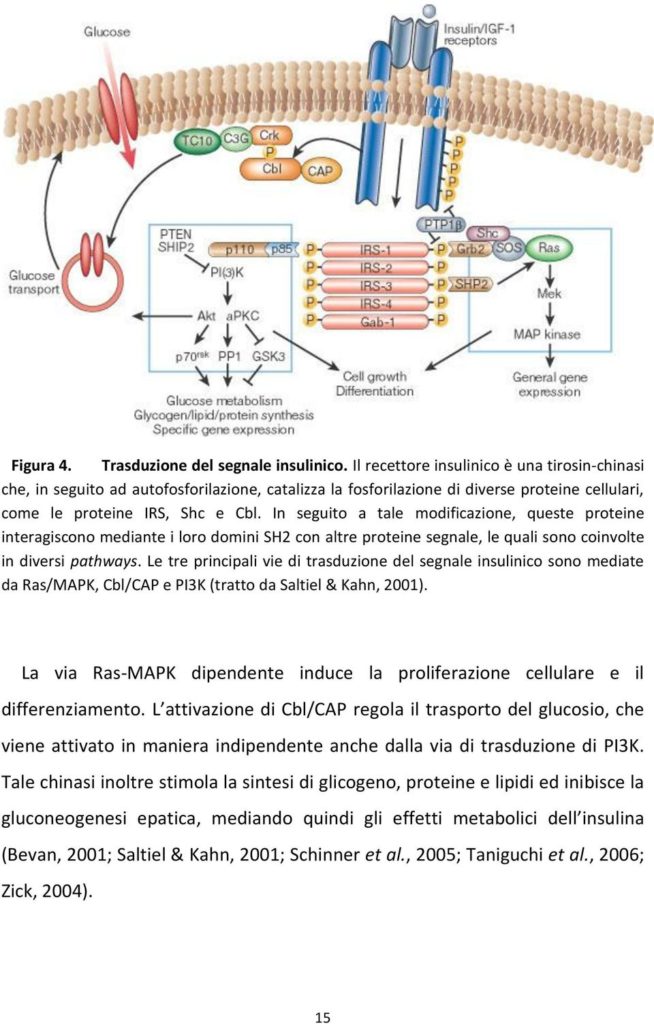
Figura 1 – Trasduzione del segnale insulinico (tratta da Saltiel & Kahn, 2001).
Parole chiave: Infiammazione, azione insulinica, tessuto adiposo viscerale, recettori toll-like.
| Indirizzo per la corrispondenza Anna Solini, MD PhD Dipartimento di Medicina Interna Università di Pisa – Via Roma, 67 – 56100 Pisa E-mail: anna.solini@med.unipi.it | |
| Introduzione L’insulina è l’ormone chiave nella regolazione del metabolismo glicolipidico. L’insulino resistenza può essere definita come una mancata o ridotta risposta tissutale agli effetti biologici dell’ormone. Si tratta, generalmente, di un fenomeno post-recettoriale dovuto ad un difetto della cellula bersaglio. La trasduzione del segnale insulinico avviene principalmente attraverso tre vie intracellulari, mediate da MAPK (Mitogen-Activated Protein Ki- nase), Cbl/CAP (Cbl-associated protein) o PI3K (Phosphatidylinositol 3-kinase) (1) (Figura 1). L’insulino resistenza nel fegato e nel muscolo scheletrico, insieme all’alterazione anatomo-funzionale della β cellula, configura il difetto fisiopatologico che è alla base del diabete tipo 2, alla cui complessa patogenesi concorrono comunque anche il tessuto adiposo (attraverso una aumentata lipolisi), il tratto gastrointestinale (attraverso un difetto dell’asse incretinico), la cellula (iperglucagonemia), il rene (aumento della gluconeogenesi ed aumentato riassorbimento tubulare del glucosio) e il cervello (insulino resistenza) (2-4). |
La resistenza all’insulina non è comunque appannaggio esclusivo del paziente diabetico, in quanto un certo grado di insulino resistenza si riscontra comunemente in altre condizioni cliniche quali l’iper tensione, la dislipidemia e, soprattutto, l’obesità. Quest’ultima condizione, risultato dell’eccessivo introito calorico e della inattività fisica, gioca un ruolo chiave nello sviluppo della insulino resistenza e della disfunzione β pancreatica (5, 6).
Un aumentato deposito di trigliceridi, specialmente se localizzato a livello viscerale, porta alla formazione di adipociti voluminosi che risultano particolarmente resistenti all’effetto antilipolitico dell’insulina, e che quindi rilasciano grandi quantità di acidi grassi liberi (FFA) e glicerolo.
Questo particolare fenotipo, caratterizzato da un aumento dei trigliceridi e delle LDL ossidate, insieme a ridotti livelli di HDL, è responsabile del profilo di lipotossicità del diabete (7).
Lipotossicità è un termine usato per de- scrivere l’effetto deleterio dell’accumulo tissutale di grasso sul metabolismo del glucosio, ed include il concetto che un aumento dei livelli plasmatici ed intramiocellulari di metaboliti lipidici tossici (come acil-CoA a lunga catena, diacilgliceroli e ceramidi) svolgano un ruolo preponderante nella patogenesi della insulino resistenza muscolare ed epatica (8). Inoltre, gli adipociti producono adipocitochine, che raggiungono distretti a distanza (quali il muscolo, il fegato e le arterie), dove esercitano ef fetti deleteri sul metabolismo e sulla funzione vascolare (9-11).
Evidenze degli ultimi 15 anni hanno documentato come l’infiammazione sia uno dei meccanismi cruciali nello sviluppo dell’insulino resistenza associata a malattie metaboliche quali l’obesità e il diabete tipo 2 (12, 13). Specificamente, è stato dimostrato che:
1) un eccessivo deposito di tessuto adi- poso, specie nel distretto viscerale, si caratterizza per una aumentata espressione e rilascio di citochine pro-infiammatorie quali il tumor necrosis factor (TNF)-a e la interleuchina-6 (IL-6);
2) questo eccesso di tessuto adiposo atti- va vie di segnale infiammatorie come risultato di una disregolazione cellulare dei pathways omeostatici, come la risposta allo stress del reticolo endo- plasmico;
3) gli FFA, i cui livelli aumentano nel corso di diverse malattie metaboliche, possono attivare pathways pro-infiammatori, portando allo sviluppo dell’insulino resistenza.
Meccanismi attraverso cui l’infiammazione può indurre insulino resistenza
Lipotossicità
Lipotossicità e infiammazione sono intimamente coinvolti nella evoluzione di diverse malattie metaboliche. L’eccessivo accumulo di lipidi all’interno dei macrofa- gi nell’endotelio vascolare induce la loro trasformazione fenotipica in foam cells, secrezione di citochine proinfiammatorie ed apoptosi, contribuendo alla progressione dell’aterosclerosi. Nel fegato steatosico, l’attivazione delle cellule di Kupf fer e i macrofagi residenti possono favorire l’infiammazione del fegato e la sua evo-
luzione in steatoepatite (14). L’accumulo di lipoproteine e derivati lipidici nel rene induce infiltrazione macrofagica e attiva il danno infiammatorio tipico delle malattie renali croniche.
Alla luce di queste semplici considerazioni, appare ovvio come un eccesso di tessuto adiposo configuri uno stato di infiammazione sub-clinica. Il tessuto adiposo, quando rappresentato in quantità normale, contiene una popolazione quiescente di macrofagi locali con scarsissima attività pro-infiammatoria, che ha il compito di sostenere la funzione adipocitaria e mantenere una normale sensibilità insuli- nica tissutale.
Quando gli adipociti si ipertrofizzano, secernono una quantità eccessiva di fattori chemiotattici che promuovono l’infiltrazione macrofagica (15); altri macrofagi possono essere reclutati semplicemente attraverso l’aumento delle cellule necrotiche, più rappresentate nel tessuto adiposo dell’obeso.
I macrofagi che infiltrano il tessuto adiposo sono anche caratterizzati da fenotipi diversi: quelli cosiddetti “residenti” (M2) esprimono la proteina chitinasi-like Ym1, l’arginasi (un enzima che blocca l’attività della NO sintasi inducibile (iNOS), e producono citochine anti-infiammatorie, quali IL-10, mentre i macrofagi “reclutati” (M1) esprimono il marker di superficie CD11c ed iNOS, e producono alti livelli di citochine pro-infiammatorie, quali IL-6 e TNFa (16). Inoltre, la fuoriuscita di FFA da questi grossi adipociti è in grado di attivare il signaling del Toll-like receptor (TLR) e, a valle, le vie di segnale Janus N-terminal kinase (JNK) e nuclear factor kB (NF-kB) nei macrofagi residenti, inducendo anche attraverso queste vie un deciso viraggio verso un classico fenotipo pro-infiammatorio (Figura 2).
Diretta conseguenza di ciò è l’aumentata produzione di citochine, tra cui IL-6 e TNFa, che interferiscono con la nor- male trasmissione del segnale insulinico favorendo l’insorgenza del diabete tipo 2 e propagando ulteriormente lo stato di infiammazione cronica (17, 18).
Quindi, la comunicazione tra adipociti e macrofagi è impor tante nell’innescare la risposta infiammatoria adipocitaria ed entrambi i tipi cellulari contribuiscono
all’insorgenza della insulino resistenza; infatti, sebbene i macrofagi infiltranti sia- no la fonte principale di TNFa, anche gli adipociti sono fonte di una quantificabile frazione della IL-6 circolante nei pazienti obesi.
È importante anche sottolineare come l’obesità si caratterizzi non solo per la re- sistenza agli ef fetti biologici dell’insulina, ma anche a quelli della leptina, sia nei tessuti periferici che a livello del sistema ner- voso centrale (19).
Figura 2 – Cross-talk tra adipociti e macrofagi infiltranti nel tessuto adiposo.
In linea con questo concetto, studi recenti indicano che l’assunzione di una dieta ad alto contenuto di grassi induce infiammazione in alcune regioni chiave del sistema nervoso che controllano l’omeostasi energetica e il metabolismo periferico del glucosio.
Stress del Reticolo Endoplasmatico
Negli organismi eucarioti tutte le proteine secretorie passano nel reticolo endoplasmatico (ER), dove vengono ripiegate assumendo la loro struttura tridimensionale, in un processo detto folding, ed assemblate in complessi, prima di essere traspor tate nel Golgi.
L’ER è molto sensibile all’eccesso di glucosio, di lipidi ed ai patogeni, per cui può essere considerato un sito d’integrazione tra la risposta immune e quella me- tabolica.
È stato suggerito che l’ER sia responsabile della percezione dello stress metabolico e della conseguente induzione di una risposta infiammatoria; primo responsabile di questo stress sarebbe un eccessivo accumulo di tessuto adiposo (20, 21). Il conseguente accumulo di proteine non ripiegate compor ta l’attivazione di una specifica risposta, denominata UPR (unfolded protein response), atta a ristabilire l’integrità funzionale di tale organello citoplasmatico. Se il difetto di folding proteico non è risolto, si realizza un’attivazione cronica di UPR, la quale induce l’apoptosi (22).
Figura 3 – Elevati livelli di glucosio ed acidi grassi liberi inducono stress ossidativo e del reticolo endopla- smico, con conseguente danno della β cellula.
Inoltre lo stress dell’ER è coinvolto nell’induzione di insulino resistenza, tramite l’attivazione di JNK e dell’inibitore del fattore nucleare kappa-B subunità beta (IKK-β), l’induzione di Tribbles (TRB)3, una pseudochinasi intracellulare che modula l’attività di numerose vie di trasduzione del segnale, e la produzione di specie reattive dell’ossigeno (ROS) (23, 24).
Stress ossidativo
È stato dimostrato che i ROS sono prodotti in diversi tessuti attraverso vari meccanismi quali reazioni di glicosilazione non enzimatica, traspor to mitocondriale di elettroni ed aumentata attività della nicotinamide adenin dinucleotide fosfato (NADPH) ossidasi. Varie alterazioni metaboliche, tra cui l’iperglicemia nel paziente diabetico o l’aumento degli FFA che si riscontra nell’obesità, possono indurre stress ossidativo attraverso una aumentata attività mitocondriale (aumentato disaccoppiamento mitocondriale e βossidazione), con conseguente aumentata produzione di ROS. Anche il TNFa sembra avere un effetto nell’induzione dei ROS, che a loro volta stimolano la produzione di tale citochina (25, 26).
I ROS sono anche modulatori negativi della azione insulinica, interferendo con la redistribuzione cellulare insulinoindotta dell’insulin receptor substrate-1 (IRS-1) e PI3K, compromettendo così la traslocazione del traspor tatore del glucosio GLUT4; infine accelerano la degradazione di NO, riducendone la biodisponibilità e compromettendone le proprietà antiossidanti, meccanismi che concorrono anch’essi al determinismo della insulino resistenza (27).
Una cellula particolarmente sensibile al danno da ROS è la β cellula pancreatica (Figura 3), perchè relativamente povera di enzimi antiossidanti quali la catalasi, la glutatione perossidasi e la superossido dismutasi, in grado di contrastare l’azione dei radicali liberi. I ROS possono anche danneggiare indirettamente la cellula attraverso una serie di vie di segnale intra- cellulari sensibili allo stress, tra cui Nuclear factor-kB (NF-kB), mitogen-activated protein kinase p38 (p38MAPK), kinases JunNH2-terminal/kinases of proteins activated by stress (JNK/SAPK), esosamine, protein chinasi C (PKC) e via dei polioli(28).
Tra queste vie di segnale, la pathway NF-kB gioca un ruolo chiave come intermediatore di risposte immuni ed infiammatorie, regolando l’espressione di numerosi geni. Essendo un pathway intracellulare target dell’iperglicemia e dei ROS, la sua attivazione può anche essere indotta da stimoli endogeni ed esogeni, tra cui un eccesso di FFA, TNFa, interleuchina 1β (IL-1β) e altre citochine proinfiammatorie, prodotti di glicosilazione avanzata (AGE) e loro recettori (RAGE), p38MAPK, danno al DNA genomico, infezioni virali e radiazioni ultraviolette.
Disfunzione mitocondriale
I soggetti insulino resistenti, specie se obesi e/o affetti da diabete tipo 2, sono caratterizzati da una ridotta espressione di Peroxisome proliferator-activated receptor gamma coactivator-1c (PGC-1) e Nuclear respirator y factor-1 (NRF-1) nel muscolo scheletrico, cui si associa una ridotta ossidazione degli acidi grassi e una ridotta biogenesi mitocondriale (29, 30).
Tale fenomeno può essere correlato ad una scarsa attività fisica, ad una dieta ricca di grassi, oppure a mutazioni nel DNA mitocondriale (mtDNA) correlate all’invecchiamento, nonché alla presenza di un polimorfismo genetico di PGC-1 (31).
Altri mediatori di infiammazione
Oltre alle vie di segnale dipendenti dalle serin/treonin chinasi, diversi altri fattori contribuiscono all’insulino resistenza indotta da infiammazione; tra questi vale la pena citare le proteine SOCS (suppressor of cytokine signaling), la cui attivazione è favorita da diverse citochine infiammatorie, quali TNFa e IL-1β. SOCS-1, -3 e -6 sono coinvolte nell’inibizione della fosforilazione in tirosina di IRS-1 e -2 o nella loro degradazione ad opera dei proteosomi.
SOCS-1 e -3 possono favorire l’insulino resistenza attraverso l’attivazione di Ste- rol regulator y element binding protein-1c (SREBP-1c) (32). È comunque interessante segnalare che, in un modello murino, è stato recentemente riportato come l’espressione di SOCS-1 nelle cellule ematopoietiche protegga dalla infiammazione sistemica e dalla insulino resistenza epatica (33).
TLRs e immunità innata
I TLRs sono pattern-recognition receptors (PRRs) e, nel sistema immune innato, svolgono il compito fondamentale di rivelare la presenza e la natura di una infezio- ne microbica, garantendo la prima linea di difesa dell’ospite.
I TLRs (ad oggi ne sono stati identificati 12 nei mammiferi) comprendono una famiglia di recettori transmembrana di tipo I, caratterizzati da un dominio extracellulare Leucine Rich Repeat (LRR) e un dominio intracellulare recettore Toll/IL-1
(TIR) che mostra omologia con quello del recettore di IL-1.
Il dominio TIR è necessario per l’interazione e il reclutamento di diverse “moleco- le adattatrici” in grado di attivare diverse vie di trasduzione intracellulare del segnale. I TLRs sono espressi in compartimenti cellulari distinti e riconoscono vari pattern molecolari patogeno-associati, derivati da vir us, batteri, funghi patogeni e parassiti. TLR1, TLR2, TLR4, TLR5, TLR6 e TLR11 sono espressi sulla super ficie cellulare mentre TLR3, TLR7, TLR8 e TLR9 sono espressi in compar timenti intracellulari quali gli endosomi e il reticolo endopla- smatico (34, 35).
TLR4 è presente in diversi tipi cellulari, prevalentemente in cellule del sistema immunitario, compresi macrofagi e cellule dendritiche; esso lega i lipopolisaccaridi (LPS) delle pareti cellulari dei batteri Gram-negativi. È interessante notare come acidi grassi a media catena, componenti dei LPS, siano in grado di innescare il signaling TLR4 in una linea cellulare di macrofagi (36); gli acidi grassi saturi, i cui livelli plasmatici sono spesso aumentati nell’obesità e nel diabete tipo 2, si candidano quindi a promuovere insulino resistenza anche attraverso questo meccanismo.
A conferma di questa ipotesi, l’alto glucosio e gli acidi grassi sinergizzano nel promuovere la reattività e la produzione di citochine da par te dei monociti attraverso una attivazione di questa pathway (37) ed i soggetti insulino resistenti si caratterizzano per una iperespressione di TLR4 (38).
La capacità degli acidi grassi liberi di attivare i TLRs varia in rapporto alla lun- ghezza della loro catena e correla con la loro abilità di indurre insulino resistenza nel muscolo scheletrico; l’acido palmitico (C16:0) e lo stearico (C18:0) sono potenti attivatori del signaling infiammatorio, e inducono insulino resistenza. Analisi di
espressione genica nel muscolo cardiaco, dopo il trattamento con acidi grassi saturi con catene di lunghezze differenti, mostrano come il palmitato sia in grado di indurre specificamente l’espressione di geni che codificano per proteine dello stress del reticolo endoplasmatico e dello stress ossidativo (39). In accordo con questo ruolo del palmitato, topi con una mutazione naturale o indotta del TLR4 sono protetti dall’insulino resistenza associata all’obesità (40).
TLR2 è espresso sulla superficie cellulare, ed è coinvolto nel riconoscimento di una gran varietà di componenti microbiche derivate da batteri, funghi, parassiti e virus. L’inibizione di TLR2 migliora la sen- sibilità all’insulina e la trasmissione del segnale ormonale nel muscolo e nel tessuto adiposo di topi nutriti con una dieta ad alto contenuto di grassi. Inoltre, l’inibizione attraverso RNA inter ference del TLR2 in cellule muscolari determina una inibizione pressochè completa della insulino resistenza indotta da palmitato (41).
Gli acidi grassi saturi sembrano, quindi, candidarsi a potenti mediatori di insulino resistenza anche agendo da ligandi per TLR4 e attivando le cascate del segnale JNK e IKK, che inibiscono entrambe l’azione dell’insulina nel muscolo scheletrico, nel fegato e nel tessuto adiposo. Esistono però evidenze a sostegno di un effetto diretto degli acidi grassi sul signaling TLR-dipendente, con lo sfingolipide ceramide, la cui sintesi dipende dai grassi saturi, a fare da intermediario molecolare candidato a legare gli stress metabolici, come eccessi di nutrienti e citochine infiammatorie, all’induzione dell’insulino resistenza.
Il ceramide è un regolatore onnipresente dello stress cellulare, e l’inibizione della sua biosintesi migliora considerevolmente l’omeostasi del glucosio in roditori con diabete e obesità; inoltre, una risposta pro-infiammatoria ai ligandi del TLR da par te dei monocito-macrofagi può essere amplificata attraverso un pathway separato TLR4-indipendente, che richiede il processamento metabolico degli acidi grassi saturi a ceramide (42). La insulino resistenza indotta dall’attivazione di TLR4 richiede, a monte, la biosintesi di cerami- de (43). Infine, l’attivazione del TLRs può potenzialmente avvenire attraverso un au- mento delle concentrazioni circolanti di LPS leganti il TLR4.
È stato recentemente suggerito che la flora batterica intestinale rappresenti un fattore impor tante per il metabolismo e per le riserve di energia dell’ospite e LPS è stato identificato come fattore d’innesco nelle prime fasi dello sviluppo dell’infiammazione e del deterioramento metabolico. LPS viene prodotto nell’intestino dell’ospite in seguito alla morte dei batteri gram-negativi; segue una traslocazione nei capillari intestinali attraverso un meccanismo TLR4-dipendente e un traspor to ai tessuti bersaglio, dove LPS innesca il signaling infiammatorio, legando e attivando TLR4. I grassi introdotti con la dieta aumentano la porzione di flora batterica intestinale in grado di produrre LPS (44). Questo stato, caratterizzato da una super-regolazione dell’infiammazione, viene definito endo- tossiemia metabolica, e favorisce sia l’in- cremento ponderale che la comparsa del diabete. Nonostante la vivacità della ricer- ca scientifica recente in questo ambito, il r uolo tessuto-specifico del signaling TLR- dipendente nello sviluppo della resistenza insulinica sistemica è solo in parte noto. Studi futuri dovranno chiaramente af fron- tare direttamente il ruolo del signaling TLR nei tessuti primariamente responsivi all’insulina, quali il muscolo scheletrico, il fegato e il tessuto adiposo attraverso la conduzione di studi in vivo.
Meccanismi molecolari di insulino resistenza
La interazione tra il signaling infiammatorio e la insulino resistenza sistemica è molto complessa e di difficile interpretazione, visto che, ad esempio, anche la mancanza di IL-6 e IL-18, al pari della loro eccessiva espressione, si traduce nello sviluppo di una condizione di resistenza all’azione insulinica. Le basi molecolari della alterata azione insulinica secondaria all’aumento del signaling infiammatorio coinvolgono l’inibizione di vie di segnale a valle del recettore insulinico. La stimolazione con TNFa o elevati livelli di FFA inibisce la fosforilazione in serina di IRS-1, interrompendo la trasduzione del segnale insulinico ed inducendo insulino resistenza (45). Tra le serina/treonina china- si che possono essere attivate da stimoli infiammatori o stressogeni e contribuire alla inibizione del signaling insulinico ci sono la c-Jun N-terminal kinase (JNK), la chinasi che inibisce l’NF-κB (IKK), la 5′ AMP-activated protein kinase (AMPK) e la protein chinasi C (PKC). Queste china- si promuovono anche l’espressione di altri geni infiammatori, ad esempio attraverso l’attivazione del complesso activator pro- tein (AP)-1 (46). JNK, nelle due isoforme JNK1 e JNK2, si attiva in risposta a diversi segnali, incluse le citochine pro-infiamma- torie, gli FFA, lo stress del reticolo endoplasmatico e i ROS.
La chinasi infiammatoria IKK-β contribuisce alla insulino resistenza attraverso la fosforilazione diretta in serina di IRS-1 e la fosforilazione dell’inibitore di NF-κB (IκB), attivando quindi NF-κB e inducendo la produzione di diversi mediatori infiammatori, tra cui TNFa e IL-6. Per quanto riguarda AMPK, essa è una serina/ treonina chinasi che rifornisce il muscolo scheletrico di substrati addizionali per la produzione di energia. La sua attivazione riduce l’accumulo di tessuto adiposo intra- addominale e migliora il profilo lipidico e i livelli plasmatici di glucosio; inoltre, come documentato recentemente, l’attivazione delle sue vie di trascrizione riduce la pro- duzione di molecole infiammatorie e di ROS (47).
È impor tante sottolineare come sia inter venti farmacologici che genetici in grado di inter ferire con queste pathways possono migliorare il controllo metaboli- co, soprattutto in corso di obesità.
Conclusioni
Il concetto che l’obesità, la dislipidemia, il diabete tipo 2 siano associate ad uno stato infiammatorio subclinico cronico, e che l’attivazione di alcune vie di segnale dell’infiammazione contribuisca all’instaurarsi di una condizione di insulino resistenza, denominatore comune di queste malattie metaboliche, ha rivoluzionato la nostra conoscenza della fisiopatologia di queste malattie, la cui prevalenza è in aumento e che costituiscono un problema socioeconomico di rilevanza mondiale.
Sebbene la identificazione di alcune vie di segnale intracellulari, quali quelle mediate dalle chinasi dello stress JNK e IKK configuri nuovi e promettenti target terapeutici nel trattamento della insulino resistenza associata all’obesità, la nostra conoscenza di questo complesso scenario è tutt’altro che completa.
Evidenze recenti suggeriscono un ruolo rilevante del signaling TLRdipendente nel determinismo della insulino resistenza periferica, e la comprensione più dettagliata di questi meccanismi di attivazione dei TLRs, soprattutto in corso di obesità, rappresenta una nuova sfida per lo sviluppo di approcci innovativi nel trattamento della sindrome metabolica.
Glossario
AGE Advanced Glycosylation End Product
AMPK AMP-activated Protein Kinase
AP Activator Protein
Cbl/CAP Cbl-associated protein
ER Endoplasmic Reticulum
FFA Free Fatty Acid
GLUT4 GLUcose Transporter 4
IKK-β Inibitor of KB Kinase-βÂ
IL Interleukin
iNOS Inducible Nitric Oxide Synthase
IRS-1 Insulin Receptor Substrate-1
JNK Janus N-terminal Kinase
JNK/SAPK Kinases JunNH2- Terminal/Stress Activated Proteins Kinases
LPS Lipopolisaccaride
LRR Leucine Rich Repeat
MAPK Mitogen-Activated Protein Kinase
mtDNA Mitochondrial DNA
NADPH Nicotinamide Adenine Dinucleotide Phosphate
NF-kB Nuclear factor-kB
NRF-1 Nuclear Respiratory Factor-1
p38MAPK p38 Mitogen-Activated Protein Kinase
PGC-1: Peroxisome proliferatoractivated receptor gamma coactivator-1
PI3K Phosphatidyl-inositol 3-kinase
Infiammazione sistemica ed insulino resistenza
Bibliografia
- Saltiel Ar, Kahn CR. Insulin signaling and the regulation of glucose and lipid metaboli- sm. Nature. 2001; 414: 799-806.
- DeFronzo RA. Insulin resistance, lipotoxici- ty, type 2 diabetes and atherosclerosis: the missing links. The Claude Bernard Lecture 2009. Diabetologia. 2010; 53: 1270-1287.
- Weir GC, Marselli L, Marchetti P, et al. To- wards better understanding of the contri- butions of overwork and glucotoxicity to the beta-cell inadequacy of type 2 diabetes. Diabetes Obes Metab. 2009; 11 (Suppl. 4): 82-90.
- Drucker DJ. The role of gut hormones in glucose homeostasis. J Clin Invest. 2007; 117: 24-32.
- Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Dia- betes Obes. 2011; 18: 139-143.
- Ferrannini E, Camastra S, Gastaldelli A et al. Beta-cell function in obesity: effects of weight loss. Diabetes. 2004; 53 (Suppl. 3): S26-S33.
- Carpentier AC. Postprandial fatty acid me- tabolism in the development of lipotoxicity and type 2 diabetes. Diabetes Metab. 2008; 34: 97-107.
- Amati F, Dubé JJ, Alvarez-Carnero E et al. Skeletal muscle triglycerides, diacylglyce- rols, and ceramides in insulin resistance: another paradox in endurance-trained athle- tes? Diabetes. 2011; 60: 2588-2597.
- Deng Y, Scherer PE. Adipokines as novel biomarkers and regulators of the metabolic syndrome. Ann NY Acad Sci. 2010; 1212: E1- E19.
10.Kawamoto R, Tabara Y, Kohara K, et al. Serum high molecular weight adiponectin correlates with arterial stiffness in community-dwelling persons. Endocr Res. 2011; 36: 53-63.
- Costandi J, Melone M, Zhao A, Rashid S. Human resistin stimulates hepatic over- production of atherogenic ApoB-containing lipoprotein particles by enhancing ApoB stability and impairing intracellular insulin signaling. Circ Res. 2011; 108: 727-742.
- Mraz M, Lacinova Z, Drapalova J, et al. The
Anna Solini, Edoardo Vitolo, Mario luca Morieri, Giovanni zuliani effect of ver y-low-calorie diet on mRNA expression of inflammationrelated genes in subcutaneous adipose tissue and peripheral monocytes of obese patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2011; 96: E606-613.
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immu- nol. 2011; 11: 98-107.
- Perseghin G. Lipids in the wrong place: vi- sceral fat and nonalcoholic steatohepatitis. Diabetes Care. 2011; 34 (Suppl. 2): S367- S370.
- Suganami T, Ogawa Y. Adipose tissue ma- crophages: their role in adipose tissue re- modeling. J Leukoc Biol. 2010; 88: 33-39.
- Hirata Y, Tabata M, Kurobe H, et al. Coro- nar y atherosclerosis is associated with ma- crophage polarization in epicardial adipose tissue. J Am Coll Cardiol. 2011; 58: 248-255.
- Tilg H, Moschen AR. Inflammatory mechanisms in the regulation of insulin resistance. Mol Med. 2008; 14: 222-231.
- Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011; 13: 11-22.
- Wauman J, Tavernier J. Leptin receptor signaling: pathways to leptin resistance. Front Biosci. 2011; 17: 2771-2793.
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006; 444: 860-867.
- Birkenfeld AL, Lee HY, Majumdar S, et al.Influence of the hepatic eukar yotic initiation factor 2{alpha} (eIF2{alpha}) endoplasmic reticulum (ER) stress response pathway on insulin-mediated ER stress and hepatic and peripheral glucose metabolism. J Biol Chem. 2011; 286: 36163-36170.
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticu- lum stress. Nat Cell Biol. 2011; 13: 184-190.
- Ozcan U, Cao Q, Yilmaz E et al. Endopla- smic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004; 306: 457-461.
- Malhotra JD, Kaufman RJ. Endoplasmatic reticulum stress and oxidative stress: a vi- cious cycle or a double-edged sword? An- tiox Redox Signal. 2007; 9: 2277-2293.
- Rains JL, Jain SR. Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med. 2011; 50: 567-575.
- 26. Morgan MJ, Liu ZG. Reactive oxygen species in TNFalpha-induced signaling and cell death. Mol Cells. 2010; 30(1): 1-12.
- 27. Potenza MA, Gagliardi S, Nacci C, et al. Endothelial dysfunction in diabetes: from mechanisms to therapeutic targets. Curr Med Chem. 2009; 16: 94-112.
- 28. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011; 21: 103-115.
- 29. Wang CH, Wang CC, Wei YH. Mitochondrial dysfunction in insulin insensitivity: implication of mitochondrial role in type 2 diabetes. Ann N Y Acad Sci. 2010; 1201: 157-165.
30. Patti ME, Butte AJ, Crunkhorn S, et al. Co- ordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003; 100: 8466-8471.
31. Hara K, Tobe K, Okada T, et al. A genetic variation in the PGC-1 gene could confer in- sulin resistance and susceptibility to Type II diabetes. Diabetologia. 2002; 45: 740-743.
32. Ueki K, Kondo T, Tseng YH, et al. Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc Natl Acad Sci USA. 2004; 101: 10422-10427.
33. Sachithanandan N, Graham KL, Galic S, et al. Macrophage deletion of SOCS1 increa- ses sensitivity to LPS and palmitic acid and results in systemic inflammation and hepa- tic insulin resistance. Diabetes. 2011; 60: 2023-2031.
34. Kang JY, Lee JO. Structural biology of the Toll-like receptor family. Annu Rev Bio- chem. 2011; 80: 917-941.
35. Kawai T, Akira S. Signaling to NF-κB by Toll-like receptors. Trends Mol Med. 2007; 13: 460–469.
36. Wong SW, Kwon MJ, Choi AM, et al. Fatty acids modulate Toll-like receptor 4 activa- tion through regulation of receptor dimeri- zation and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem. 2009; 284: 27384-27392.
37. Dasu M, Jalal I. Free fatty acids in the pre- sence of high glucose amplify monocyte inflammation via Toll-like receptors. Am J Physiol Endocrinol Metab. 2011; 300: E145- E154.
38.Reyna SM, Ghosh S, Tantiwong P, et al. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes. 2008; 57: 2595-2602.
- Lockridge JB, Sailors ML, Durgan DJ, et al. Bioinformatic profiling of the transcrip- tional response of adult rat cardiomyocytes to distinct fatty acids. J Lipid Res. 2008; 49: 1395-1408.
- Tsukumo DM, Car valho-Filho MA, Car val- heira JB, et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007; 56: 1986-1998.
- Senn JJ. Toll-like receptor-2 is essential for the development of palmitate-induced in- sulin resistance in myotubes. J Biol Chem. 2006; 281: 26865-26875.
- Li X, Becker KA, Zhang Y. Ceramide in re- dox signaling and cardiovascular diseases. Cell Physiol Biochem. 2010; 26: 41-48.
43. Holland WL, Bikman BT, Wang LP, et al. Lipid-induced insulin resistance mediated by the proinflammator y receptor TLR4 re- quires saturated fatty acid-induced cerami- de biosynthesis in mice. J Clin Invest. 2011; 121: 1858-1870.
44. de La Serre CB, Ellis CL, Lee J, et al. Pro- pensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Phy- siol Gastrointest Liver Physiol. 2010; 299: G440-G448.
45. Grounds MD, Radley HG, Gebski BL, et al. Implications of cross-talk between tumour necrosis factor and insulin-like growth fac- tor-1 signaling in skeletal muscle. Clin Exp Pharmacol Physiol. 2008; 35: 846-851.
46. Fan D, Li L, Wang C, et al. Adiponectin indu- ces interleukin-6 production and its under- lying mechanism in adult rat cardiac fibro- blasts. J Cell Physiol. 2011; 226: 1793-1802.
47. Salminen A, Hyttinen JM, Kaarniranta K. AMP-activated protein kinase inhibits NF- κB signaling and inflammation: impact on healthspan and lifespan. J Mol Med. 2011; 89: 667-676.